
Fitness
Early Release – Loop-Mediated Isothermal Amplification Assay to Detect Invasive Malaria Vector Anopheles stephensi Mosquitoes – Volume 30, Number 9—September 2024 – Emerging Infectious Diseases journal
Disclaimer: Early release articles are not considered as final versions. Any changes will be reflected in the online version in the month the article is officially released.
Author affiliations: US President’s Malaria Initiative, Centers for Disease Control and Prevention, Atlanta, Georgia, USA (C. Rafferty, S. Zohdy); Centers for Disease Control and Prevention, Atlanta (C. Rafferty, G. Raise, J. Scaife, S. Zohdy); Kenya Medical Research Institute (KEMRI), Kisumu, Kenya (B. Abongo, S. Omondi, S. Milanoi, M. Muchoki, B. Onyango, E. Ochomo)
In 2012, Anopheles stephensi, a primary malaria vector in South Asia, was detected in Djibouti, a country in Africa that was approaching malaria preelimination status (1). Unlike typical malaria vectors in Africa, An. stephensi mosquitoes can thrive in both urban and rural environments. After the detection in Djibouti, An. stephensi mosquitoes were reported in Ethiopia and Sudan in 2016, Somalia in 2019, Nigeria in 2020, Kenya in 2022, and Ghana and Eritrea in 2023 (2). The initial detection in Djibouti came during a malaria outbreak (1), after which a 36-fold increase in malaria was reported from 2012 to 2020 (3). In Dire Dawa, the second largest city in Ethiopia, an unusual dry season outbreak of malaria was reported in 2022, and epidemiologic and entomologic investigations incriminated An. stephensi mosquitoes as driving the outbreak (4). Furthermore, the species’ insecticide resistance status and unique bionomics present a challenge to proven malaria vector control tools, such as insecticide-treated bed nets and indoor residual spraying (5,6). Modeling studies have predicted that if An. stephensi mosquitoes continue to spread throughout Africa, an additional 126 million persons, predominantly in urban areas, will be at risk for malaria (7,8). To respond to this threat, the World Health Organization (WHO) launched an initiative to halt the spread of An. stephensi mosquitoes (9), and global organizations (10) and countries have released action plans to encourage enhanced surveillance for the species for early detection in new locations and rapid response to halt spread and mitigate impacts.
Despite efforts to enhance surveillance for An. stephensi mosquitoes in Africa, the species was not included in morphologic keys until 2020 (11). Therefore, the mosquitoes be missed in routine surveillance activities, and An. stephensi mosquitoes could be misidentified as the more common malaria vector An. gambiae sensu lato if morphological identification is inadequate (3). In addition, reporting a detection of An. stephensi mosquitoes in a new country to WHO requires molecular confirmation, which can be challenging in resource-limited settings. Surveillance for An. stephensi mosquitoes often requires larval surveys (12) because routine malaria vector adult collections are not optimal for the species (6) and currently a validated key to identify An. stephensi larvae is not available, so larval samples that do not emerge to adults may also require molecular confirmation.
In 2023, a PCR protocol for An. stephensi species identification was released and shown to detect An. stephensi mosquitoes even among pooled samples, presenting a promising avenue for molecular detection (13). However, PCR can be time consuming and limited by molecular laboratory capacity, access to reagents, trained personnel, and assay specificity and interpretation.
Loop-mediated isothermal amplification (LAMP) assays have been used since the 1990s for rapid amplification of gene targets (14), resulting in a visual change through fluorescence, turbidity, or color that provides a qualitative indicator of positivity. In this way, LAMP assays function like conventional PCRs, which yield a band (positive) or no band (negative). However, instead of requiring temperature cycling like PCR, LAMP assays produce copies through looped primer sets at 1 consistent temperature, removing the need for a thermal cycler and instead requiring only a heat block, water bath, or any other device that keeps temperature constant. One study even used hand-warmers and a Styrofoam cup to conduct a LAMP assay (15). Because the COVID-19 pandemic increased the need for rapid diagnostics, LAMP technology evolved to include colorimetric and dipstick assays (16).
To address the challenges that invasive An. stephensi mosquito surveillance and corresponding molecular confirmation present, the aim of this study was to develop an easy-to-interpret, rapid colorimetric LAMP-based Anopheles stephensi species (CLASS) identification assay, specifically designed and optimized for use in resource-limited settings or for rapid high-throughput screening. To ensure accuracy and feasibility for deployment of the developed assay, we sought to design optimal primers and assay conditions, determine assay sensitivity and pooling strategies, determine assay specificity when compared with congeners or conspecifics, develop direct sample amplification approaches without the need for DNA extraction, compare results between the existing PCR protocol and CLASS assay, and evaluate CLASS on wild-caught, sequence-confirmed invasive An. stephensi mosquitoes from Kenya.
LAMP Primer Design and Optimization
We designed the LAMP primers by using the NEB LAMP Primer Design Tool version 1.4.1 (New England Biolabs, https://www.neb.com) (17). We used the internal transcribed spacer 2 rDNA region unique to An. stephensi species, using a sequence from GenBank (accession no. MW732931.1) (18). One LAMP primer set contains 5 primers as follows: an outer forward primer (F3), an inner forward primer (FIP), an outer backward primer (B3), an inner backward primer (BIP), and a loop primer (Figure 1) (19,20). Attempts to set fixed primers resulted in no possible loop primer combinations by the program; therefore, we used default parameters and allowed the program to choose primers.
Of 4 possible primer sets, 2 contained primers in the species-specific region. We tested those primers across a temperature gradient using 2 sets of differing concentrations. Initial test concentrations were adapted from NEB kit manufacturer recommendations, and primer concentrations were based on an An. gambiae species identification LAMP assay (21). One primer set showed positive, consistent results and minimum cross-reactivity to other species. We then further optimized that primer set for maximum specificity (Table 1; Figure 1).
Insectary-Reared An. stephensi and Other Mosquito Species
We obtained larvae and adult insectary-reared and maintained colony mosquitoes from 8 distinct non–An. stephensi Anopheles species, 3 strains of An. stephensi mosquitoes of different origins (STE2 from India, SDA 500 from Pakistan, UCI from India), and one Aedes (Ae. aegypti) mosquito. Mosquitoes came from the Malaria Research and Reference Reagent Resource Center through BEI Resources (Table 2) (22).
DNA Extraction
We extracted DNA from whole adult, single mosquito leg, and whole 3rd instar larva by using the Extracta DNA Prep for PCR Kit (Quantabio Beverly, https://www.quantabio.com), adapted as follows: mosquito material added to a tube containing either 25 µL (for a single leg) or 50 µL (for a whole adult or larva) of Quantabio Extraction Reagent and incubated at 95°C for 30 minutes. We added an equal volume of Quantabio Stabilization Buffer and stored DNA at −20°C until further analysis. For the pooled species sample, we combined 1 µL of DNA from 9 DNA extractions of non–An. stephensi mosquitoes and 1 µL of DNA extracted from An. stephensi mosquitoes (STE2) in a microfuge tube and mixed contents.
CLASS Assay
We carried out CLASS reactions by using the NEB WarmStart Colorimetric LAMP 2X Master Mix (New England Biolabs), according to manufacturer recommendations but optimized as follows: 1 µL of genomic DNA was added to 12.5 µL of WarmStart Colorimetric LAMP 2X Master Mix and 10X primers at final concentrations of 5 µM of B3 and F3 primers, 20 µM of BIP and FIP primers, and 10 µM of LF primer. We added molecular-grade water to reach a final volume of 25 µL. We placed reaction tubes in a thermal cycler at 65°C for 30 minutes and inspected visually for color change, where positive amplification appears yellow and negative remains pink. We tested primers on extracted DNA from 12 assorted insectary-reared adults and larvae, including 3 An. stephensi strains (STE2, SDA500, and UCI), 14 field-collected specimens including 3 sequence-confirmed An. stephensi mosquitoes, and DNA from pooled species. We included a no-DNA control in each run of the assay.
Analytical Sensitivity
To test analytical sensitivity, we made a serial dilution (1:10) of DNA extract from a whole UCI An. stephensi mosquito and determined starting DNA concentration by using a NanoDrop 2000c spectrophotometer (Thermo Scientific, https://www.thermofisher.com) on 1 μL of DNA extract. For each concentration, no color change (pink) indicated a negative result and color change (yellow) a positive result. We ran samples from the dilution series in triplicate with no full change detected and 2 additional dilutions to determine the sensitivity cutoff.
Specificity Determination
We tested optimized primers against 12 laboratory anopheline strains that included 3 An. stephensi strains (SDA500, STE2, UCI) and 1 Ae. aegypti strain (Table 2). We subsequently tested the assay against 96 individual mosquitoes from each An. stephensi laboratory strain and 48 An. gambiae, An. coluzzii, An. arabiensis, An. funestus, and Ae. aegypti laboratory-reared samples for specificity and cross reactivity. We ran all reactions in triplicate to generate data on cross-reactivity with other species and specificity to An. stephensi. We included 3 An. stephensi strains to determine variations in target specificity across An. stephensi mosquitoes of different origins.
Samples for LAMP Amplification
To determine whether DNA extract is needed to run the CLASS assay or if tissue (mosquito leg, full larva, full adult mosquito) or pooled DNA amplify, we inserted single legs from insectary-reared mosquitoes directly into the master mix and compared the results with DNA extracted from a single leg. Because An. stephensi samples are often collected as larvae, we also tested the assay by immersing a whole larva into the master mix and using extracted DNA from a single larva. We also partially tested eDNA by using 1 µL using larval pan water in lieu of extracted DNA. We further tested the assay against whole adult mosquitoes and compared results with whole adult mosquito DNA. In addition, we tested pooled DNA extract from whole adult mosquitoes and from individual legs from 9 mosquito strains and 1 An. stephensi strain.
Conventional PCR Comparison
We compared CLASS results with those from a conventional An. stephensi species identification PCR assay by using previously described methods (13). We adapted the method as follows: 2X Quantabio Accustart PCR mix, 10 µM of each primer, molecular water to reach a final volume of 20 µL, and 1 µL of the extracted DNA from same species used in the CLASS assay.
CLASS Assay Validation on An. stephensi Mosquitoes from Kenya
We ran sequence-confirmed DNA extracted from wild-caught An. stephensi mosquitoes from Kenya (GenBank accession nos. OQ275144–6 and OQ878216–8) using the CLASS assay (23). We additionally tested 55 wild-caught samples collected from Marsabit, Kenya, in 2023 that previously failed to amplify via conventional PCR (24). DNA extracted at the Kenya laboratory was dried and shipped to the US Centers for Disease Control and Prevention (Atlanta, Georgia, USA), where samples were resuspended in 25 µL of PCR-grade water and stored at −20°C until processed.
LAMP Primer Design and Assay Optimization
We tested the 4 primer sets suggested by the NEB LAMP Primer Design Tool version 1.4.1 against extracted DNA from 3 An. stephensi insectary strains and 8 other Anopheles species: An. gambiae s.s., An. coluzzii, An. arabiensis, An. gambiae/coluzzii hybrid, An. funestus, An. quadriannulatus, An. dirus, and An. merus (17). We tested the 2 primer sets (P2L-45 and P26L2) that showed color change for An. stephensi samples and minimum cross-reactivity among other species with varying concentrations at 3 incubation times: 15, 30, and 45 minutes. No color change was detected at 15 minutes, but specificity was affected at 45 minutes, confirming 30 minutes as the ideal assay incubation time. We analyzed 384 reactions in duplicate (768 total reactions) using 8 different primer concentration combinations, and we chose primer set P26L2 for its consistent sensitivity and specificity (Table 3). We tested the chosen primers and respective concentration combinations using a temperature gradient (57°C, 61°C, 65°C, 69°C, 73°C, and 83°C) through 176 separate reactions. Results confirmed that primer concentration combination B3 at 65°C yielded the most consistent and specific results (Table 3). Amplification occurred at 69°C and 73°C, but specificity was inconsistent. We observed no amplification at higher or lower temperatures.
Repeated time-interval testing of 384 samples with the chosen primers showed no amplification before 25 minutes, optimum amplification at 30 minutes, and a decrease of specificity after 35 minutes. Consequently, we adopted a 30-minute incubation period for the assay. Once incubation stopped (by removal from the heat source), the product and color change remained stable and unaltered at room temperature for >12 weeks.
CLASS Assay Analytical Sensitivity
To test the sensitivity of the CLASS assay, we performed serial dilutions (1:10) of initial DNA extract to a concentration of 311.6 ng, which resulted in 100% positive color change to yellow. Positive color change was repeatedly observed at >0.0003 ng; concentrations Table 4).
CLASS Assay Specificity and Cross-Reactivity
Figure 2
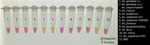
Figure 2. Visualization of testing using a colorimetric loop-mediated isothermal amplification assay to detect invasive malaria vector Anopheles stephensimosquitoes. Positive samples show a color change to yellow, whereas negative samples…
We tested the optimized P26L2 primers (Table 1) against extracted DNA from 11 insectary strains, including 3 An. stephensi mosquitoes. We ran the assay 11 separate times with different extracted DNA from single whole-colony mosquitoes for a total of 132 reactions (Figure 2). We further assessed specificity by testing DNA from 96 individual mosquitoes from each An. stephensi laboratory strain; 100% of the samples yielded a positive result. We determined cross-reactivity by sampling DNA from 48 An. gambiae, 48 An. coluzzii, 48 An. arabiensis, 48 An. funestus, and 48 Ae. aegypti whole mosquitoes and analyzing. None (0%) of the non–An. stephensi strains showed color change. We ran all specificity assays in triplicate (Table 5).
CLASS Assay Testing of Mosquito Tissue, DNA Extract, and DNA Pooling
Figure 3
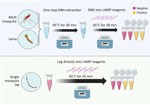
Figure 3. Schematic for colorimetric loop-mediated isothermal amplification assay to detect invasive malaria vector Anopheles stephensimosquitoes. Top: DNA from any mosquito source directly placed in the colorimetric master mix are…
Using DNA extract from a single leg resulted in color change in An. stephensi mosquitoes, with no cross-reactivity with other tested species. Inserting a single mosquito leg straight into the master mix also successfully amplified after optimization, but at a 35-minute incubation time (Figure 3). When testing a whole larva or whole adult mosquito, the assay had low specificity, and yielded cross-reactivity; however, the use of DNA extract from a single larva or mosquito from 11 Anopheles colony strains, including 3 An. stephensi strains and 1 Ae. aegypti strain, resulted in species-appropriate color change (Figure 3). Limited testing on larval pan water yielded inconclusive results. Although the CLASS assay was able to identify An. stephensi from larval pan water and not from other anopheline larval water, results showed cross-reactivity with Ae. aegypti.
CLASS Assay Specificity in Field Samples and Comparison with Conventional PCR
Figure 4

Figure 4. Validation of colorimetric loop-mediated isothermal amplification assay to detect invasive malaria vector Anopheles stephensi mosquitoes. A) Results using new assay. B) Results using existing An. stephensiPCR…
Sequence-confirmed An. stephensi samples from Kenya positively amplified using CLASS, and no cross-reactivity was seen with other Anopheles species. An. stephensi sampled in pooled DNA from 9 colony-reared species (An. gambiae s.s., An. coluzzii, An. gambiae/coluzzii hybrid, An. arabiensis, An. funestus, An. quadriannulatus, An. merus, An. dirus, and Ae. aegypti) and 1 An. stephensi sample (SDA 500) also amplified using CLASS. Conventional PCR resulted in difficult-to-interpret gel bands for An. longipalpis C, Ae. aegypti, and An. coustani samples, similar to An. stephensi samples, and inconsistently produced double bands (positive detection) on sequence-confirmed An. stephensi samples from Kenya (Figure 4, panel B).
CLASS Assay Testing of Field Samples from Marsabit, Kenya
CLASS assay testing of 55 wild-caught Anopheles samples from Marasabit, Kenya, successfully identified the 9 cytochrome c oxidase subunit I sequence-confirmed An. stephensi samples. Furthermore, no cross-reactivity was observed with the other species or unknown samples (Table 6). Twelve nonamplified samples in the sample set during barcoding also tested negative by the CLASS assay.
Molecular species identification of malaria vectors is pivotal for effective control and elimination strategies, particularly because malaria-vector mosquitoes often cannot be morphologically identified to the species level. Of increasing complexity is the introduction of invasive species, such as An. stephensi, that are not included in traditional identification keys (25) and thus can be easily misidentified. In addition, to confirm the presence of An. stephensi mosquitoes on the WHO An. stephensi Threats Map (26), molecular confirmation through Sanger sequencing (9) is required.
We developed a rapid 1-step colorimetric LAMP assay for species identification of An. stephensi mosquitoes to accelerate tracking this species across Africa or in locations where it is endemic. The CLASS identification assay provides a precise and reliable means of An. stephensi identification. Our findings indicate high sensitivity and specificity of the assay, whether An. stephensi samples were mixed in a pool of 10 other species or validated against 8 species, including 3 unique insectary-reared strains and individual wild-caught invasive An. stephensi samples. No false positives or false negatives were observed. When we conducted a dilution series to determine analytical sensitivity, even at 0.0003 ng of DNA, the CLASS assay detected An. stephensi DNA. Thus far, the specificity remains 100% when other species are processed through the assay. The ability to differentiate between various Anopheles species, especially those with differing vectorial capacities or behaviors, is indispensable for tailoring interventions to specific vector populations (27).
The CLASS assay can be run using a single mosquito leg or DNA extract from an adult or larval mosquito (Figure 3). DNA extracted from a leg is preferable so specimens can remain largely intact for further curation, sequencing, and storage. The use of an entire mosquito or larva is highly discouraged because it yields nonspecific results and prevents further follow-up and species confirmation. For WHO submission and confirmation, sequencing-positive specimens are still encouraged; however, the CLASS assay provides a rapid, high-throughput, field-friendly screening tool for an initial detection of An. stephensi mosquitoes. Although initial testing of larval pan water yielded inconclusive results, possibly because of Aedes excessive larval shedding in the water, findings suggest the potential for additional exploration using CLASS to examine environmental DNA or large pools of specimens to yield further information about potential cross-reactivity with other species in natural settings.
A conventional PCR to support molecular detection of An. stephensi mosquitoes exists (13); however, in settings where facilities and trained personnel are limited, conventional PCR can be challenging. That PCR also has multiple primers, and thus, interpreting results can be a challenge if one or both bands are absent. In our study, insectary and field Anopheles samples run through the conventional PCR showed gel bands that could be misinterpreted as false-positive or -negative. Even insectary-reared An. stephensi samples produced inconclusive results using that assay (Figure 4, panel B). External laboratories have also reported nonamplification using the assay on samples later confirmed to be An. stephensi through sequencing (23). Follow-up PCR and Sanger sequencing validation are still critical, but our findings support the need for a robust An. stephensi assay that is simple to interpret.
The CLASS assay showed promising results when field-caught samples from Kenya were tested. Although testing on those samples used DNA, this assay’s ability to test single legs without extraction and using simple equipment suggests potential for screening large numbers of wild mosquitoes in remote settings. With additional field deployment and validation, data may be generated to potentially consider the CLASS assay as a species confirmation tool if confirmed sensitivity and specificity continue to fall within an 85% CI. In addition, alternate LAMP detection chemistries using the primers we describe could be adapted to ensure assay capacity in all settings without relying on a single company and master mix and without concern for reagent quality affecting pH change.
LAMP assay technology improved because of the need for rapid cost-effective diagnostics during the COVID-19 pandemic. Because phenol-based colorimetric LAMP assays are now widely adaptable, opportunities exist beyond An. stephensi species identification, such as for An. arabiensis, a common malaria vector in Africa currently requiring PCR for species confirmation (28). In some locations, An. arabiensis mosquitoes are the primary malaria vector, and a colorimetric LAMP screening tool could be used to rapidly distinguish non–An. arabiensis samples for further molecular confirmation. The first detection of An. stephensi mosquitoes in Ethiopia occurred when An. gambiae sensu lato mosquitoes did not amplify as An. arabiensis mosquitoes and sequencing revealed invasive An. stephensi mosquitoes instead (29).
Molecular species identification provides crucial data for epidemiologic surveillance. Real-time data on vector distribution and density guide the implementation of vector control methods, such as insecticide-treated bed nets and indoor residual spraying, ensuring that resources are used effectively to curb malaria transmission (30). Early detection, assisted by rapid assays like the CLASS assay, is critical for initiating timely responses to invasive vector populations (7).
The significance of accurate molecular identification of vector species extends beyond invasive An. stephensi. Long-term research and malaria program initiatives, guided by species identification data, enable program managers and scientists to study vector biology, behavior, and genetics. Such insights are invaluable for developing effective control tools and strategies. In addition, policy formulation relies heavily on accurate surveillance data. Molecular surveillance of vectors like An. stephensi informs policy decisions at regional, national, and international levels, ensuring a coordinated and effective response to malaria (31). Accurate molecular identification not only aids in understanding the geographic distribution of vectors but also assists in predicting potential disease outbreaks, enabling public health authorities to proactively allocate resources and plan interventions (32).
In conclusion, molecular species identification of malaria vectors, particularly in the context of invasive species such as An. stephensi, is indispensable to ensure gains made in global malaria control and elimination over the last few decades are not lost. Developing rapid, cost-effective assays, such as the CLASS assay, marks a substantial advancement in the ability to detect An. stephensi mosquitoes early in new locations, enabling rapid vector control response. This assay has potential as a screening tool to monitor the spread of the vector species. This tool is field adaptable and can be used in resource-limited settings so that laboratory capacity is not a bottleneck preventing countries from detecting and reporting the presence of An. stephensi mosquitoes. By combining accurate molecular identification of An. stephensi mosquitoes with adaptive interventions, policymakers, researchers, and public health officials can work collaboratively to mitigate the effect of this invasive malaria vector and continue to work toward a malaria-free future.
Ms. Rafferty is a molecular biologist with the US President’s Malaria Initiative in the Entomology Branch, Division of Parasitic Diseases and Malaria, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA. Her work focuses on molecular approaches for vector surveillance and control.

From Stadium to Screen: How Technology is Changing Sports Viewing

20/7/2024 Horse Racing Tips and Best Bets – Flemington, Flemington Cup day

Indian tech hub Karnataka state’s move to reserve jobs for locals not finalised, chief minister says

Financial picture dramatically improves for Shamrock Rovers in 48 hours with Sinclair Armstrong deal and European win

Our fashion editor’s favourite affordable bag is 20% off today

Jay Shah’s Big Decision Hovers Over Cricket’s Associate Member Directors Election

Budget shampoo that adds MAJOR volume boost is on sale for Amazon Prime Day: ‘Your hair feels fuller and thicker’

Being active on your commute lowers risk of disease and mental health

Tiger Woods cops brutal Open schedule as full Round 1 tee times revealed
